Structural classifcation
The central functionality of StructMAn is to perform a structural classification of all queried positions.
The classification is based on the results aggregation of all mapped structures. The criteria determining the classification are the solvent accessibility of the mapped residues of the queried position and the potential of forming an interaction with other molecules including other protein chains, DNA, RNA, metal ions and low molecular weight molecules.
There are 45 classes divided into four different types of classes:
If there is no potential interaction the queried position is classified by there solvent accessible area, leading to two cases: positions classified as lying on protein Surface or positions buried inside the protein Core.
If there is one potential interaction, the queried position is classified by the type of that interaction and the distance to the interaction partner, distinguishing between normal Interactions (distance to the interaction partner < 5 Angstrom) and far Interactions (distance to the interaction partner between 5 and 8 Angstrom). Resulting in ten different singular interaction classes, two for each of the five interaction types: Protein, DNA, RNA, Metal ions, non-metal Ions, and other low molecular weight Ligands.
If a position can be placed into two classes (e.g. the amino acid is closer than 5 Angstrom to both a ligand and another protein), they can be combined to a Double interaction class. DNA and RNA, as well as Metal and other low molecular weight ligands can not be combined, leading to 8*4 = 32 possible double interactions classes.
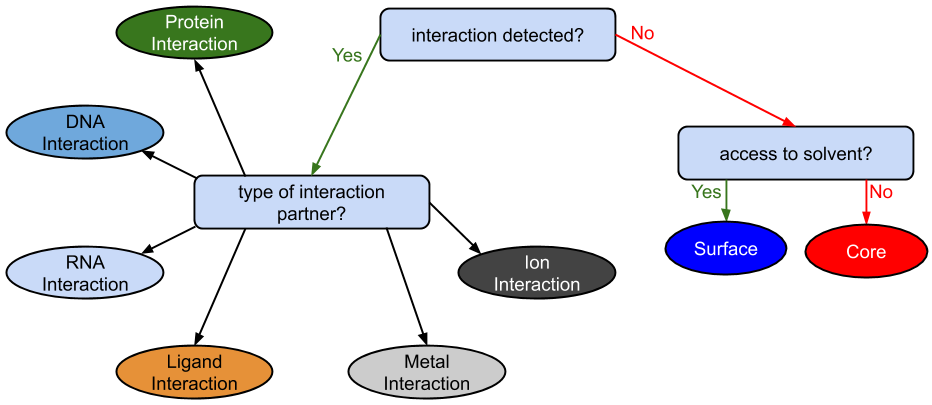 Schematic representation of the classification performed by StructMAn.
Schematic representation of the classification performed by StructMAn.